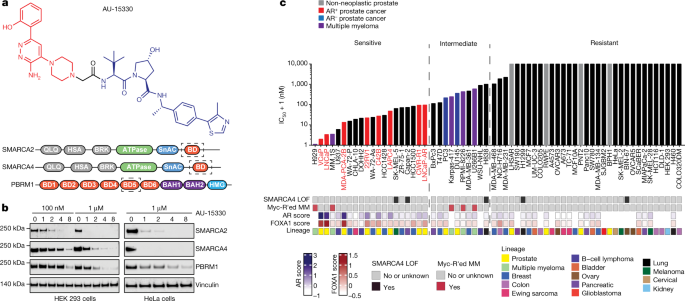
Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer Lanbo Xiao1,2 na1, Abhijit Parolia1,2,3 na1, Yuanyuan Qiao orcid.org/0000-0002-1178-34801,2,4 na1, Pushpinder Bawa orcid.org/0000-0002-8370-36381,2, Sanjana Eyunni1,2,3, Rahul Mannan orcid.org/0000-0002-6642-04681,2, Sandra E. Carson orcid.org/0000-0002-7716-96271,2, Yu Chang1,2, Xiaoju Wang1,2,4, Yuping Zhang1,2, Josh N. Vo1,2,5, Steven Kregel1,2, Stephanie A. Simko1,2, Andrew D. Delekta1,2, Mustapha Jaber1, Heng Zheng1,2, Ingrid J. Apel1,2, Lisa McMurry1,2, Fengyun Su1,2, Rui Wang1,2, Sylvia Zelenka-Wang1,2, Sanjita Sasmal6, Leena Khare orcid.org/0000-0001-9860-78876, Subhendu Mukherjee6, Chandrasekhar Abbineni6, Kiran Aithal6, Mital S. Bhakta7, Jay Ghurye7, Xuhong Cao1,2,8, Nora M. Navone orcid.org/0000-0001-8645-68909, Alexey I. Nesvizhskii orcid.org/0000-0002-2806-78191,2,4,5, Rohit Mehra orcid.org/0000-0002-6955-88841,2,4, Ulka Vaishampayan10, Marco Blanchette7, Yuzhuo Wang orcid.org/0000-0002-9749-859111,12, Susanta Samajdar6, Murali Ramachandra6 & Arul M. Chinnaiyan orcid.org/0000-0001-9282-34151,2,4,5,8,13 Nature (2021)Cite this article Chromatin remodellingProstate cancerTargeted therapies The switch/sucrose non-fermentable (SWI/SNF) complex has a crucial role in chromatin remodelling1 and is altered in over 20% of cancers2,3. Here we developed a proteolysis-targeting chimera (PROTAC) degrader of the SWI/SNF ATPase subunits, SMARCA2 and SMARCA4, called AU-15330. Androgen receptor (AR)+ forkhead box A1 (FOXA1)+ prostate cancer cells are exquisitely sensitive to dual SMARCA2 and SMARCA4 degradation relative to normal and other cancer cell lines. SWI/SNF ATPase degradation rapidly compacts cis-regulatory elements bound by transcription factors that drive prostate cancer cell proliferation, namely AR, FOXA1, ERG and MYC, which dislodges them from chromatin, disables their core enhancer circuitry, and abolishes the downstream oncogenic gene programs. SWI/SNF ATPase degradation also disrupts super-enhancer and promoter looping interactions that wire supra-physiologic expression of the AR, FOXA1 and MYC oncogenes themselves. AU-15330 induces potent inhibition of tumour growth in xenograft models of prostate cancer and synergizes with the AR antagonist enzalutamide, even inducing disease remission in castration-resistant prostate cancer (CRPC) models without toxicity. Thus, impeding SWI/SNF-mediated enhancer accessibility represents a promising therapeutic approach for enhancer-addicted cancers. In eukaryotic cells, DNA is wrapped around histone octamers (referred to as nucleosomes), which form a physical barrier to DNA-based processes4. Thus, gene expression is regulated by modifying physical accessibility of the DNA through nucleosomal remodelling and, when in an accessible state, through binding of transcription factors5,6. In this regulatory context, non-coding genomic elements called enhancers have emerged as central hubs serving as integrative platforms for transcription factor binding and activation of lineage-specific gene programs7,8. The enhancer elements can lie within untranslated or distal intergenic regions and make looping interactions with their target gene promoters to potentiate RNA polymerase II (PolII)-mediated transcription9,10.In cancer, genetic alterations invariably lead to an aberrant transcriptional state that is often wired through expansion and remodelling of the enhancer landscape11,12. This includes de novo commissioning of new enhancers (neo-enhancers) by reprogramming of pioneer factor cistromes13, enhancer hijacking via structural rearrangements14,15, and/or abnormal enhancer–promoter interactions via alterations in chromatin topology16—all to enable hyper-expression of driver oncogenes. Although there has been intense interest in therapeutically targeting aberrant enhancer function in cancer, the molecular machinery responsible for enhancer maintenance and/or activation remains poorly characterized.Recent studies have uncovered alterations in genes encoding constituent subunits of the SWI/SNF complex in over 20% of human cancers2. SWI/SNF is a multi-subunit chromatin-remodelling complex that uses energy from ATP hydrolysis to reposition or eject nucleosomes at non-coding regulatory elements, thereby enabling free DNA access for the transcriptional machinery1. In SWI/SNF-mutant tumours, the residual complex is thought to enable oncogenic transcriptional programs and speculated to be a viable therapeutic target17,18,19. Although inhibitors and degraders of ATPase and BRD7–BRD9 SWI/SNF subunits have been recently developed20,21,22, to our knowledge, no studies have comprehensively assessed the therapeutic efficacy of SWI/SNF inactivation across a wide spectrum of cancers. To this end, we have developed and characterized a highly-selective PROTAC degrader of both SWI/SNF ATPase subunits—SMARCA2 (BRM) and SMARCA4 (BRG1)—that are required for the nucleosomal-remodelling functions of SWI/SNF complexes.We found enhancer-binding transcription factor-addicted cancers (for example, AR–FOXA1-driven prostate cancer) to be exquisitely and preferentially sensitive to SWI/SNF ATPase degradation, which triggered an instantaneous, specific loss of physical accessibility and transcription factor binding at enhancer elements, thereby disrupting enhancer-wired oncogenic gene programs. To our knowledge, this study is the first preclinical proof of concept that targeted obstruction of chromatin accessibility at enhancer elements may be a potent therapeutic strategy in transcription factor-addicted tumours.We developed the PROTAC degrader, AU-15330, comprising a bait moiety that binds the bromodomain in SMARCA2 and SMARCA4 and a ligand moiety for the von Hippel–Lindau (VHL) ubiquitin ligase (Fig. 1a, Extended Data Fig. 1a). AU-15330 also binds to the secondary SWI/SNF module component PBRM1, which relies on the ATPase module for assembly onto the core complex23. Although it binds to the same bromodomain in target proteins as the PROTAC degrader ACBI120, AU-15330 comprises a distinct linker structure that largely dictates a PROTAC’s target selectivity and degradation kinetics24. Treatment of several cell lines with AU-15330 led to time and dose-dependent degradation of SMARCA2, SMARCA4 and PBRM1 (Fig. 1b). Mass spectrometry-based proteomics analysis confirmed SMARCA2, SMARCA4 and PBRM1 as the only significantly downregulated proteins (Extended Data Fig. 1b). Of note, we detected no change in the abundance of other bromodomain-containing proteins or non-targeted SWI/SNF subunits (Extended Data Fig. 1c, d). SWI/SNF complexes have been shown to assemble in a modular manner, with the ATPase module being the last to bind to the SMARCC1 (also known as BAF155)-containing core complex23. Accordingly, SMARCC1 nuclear immunoprecipitation followed by mass spectrometry showed no changes in the sequential assembly of the core and secondary modules but revealed detachment of ATPase module subunits upon AU-15330 treatment (Extended Data Fig. 1e).Fig. 1: AU-15330, a specific degrader of SWI/SNF ATPases, exhibits preferential cytotoxicity in enhancer-binding transcription factor-driven cancers.a, Structure of AU-15330 and schematic of SMARCA2, SMARCA4 and PBRM1 domains. AU-15330-targeted bromodomains (BD) are shown. QLQ, conserved Gln, Leu, Gln motif containing domain; HSA, helicase/SANT-associated domain; BRK, Brahma and Kismet domain; SnAC, Snf2 ATP coupling domain; BAH1, bromo-adjacent homology domain 1; BAH2, bromo-adjacent homology domain 2. b, Immunoblots of SMARCA2, SMARCA4 and PBRM1 on treatment of HEK 293 and HeLa cells with AU-15330 at increasing concentrations or time durations. Vinculin is used as a loading control, and is probed on a representative immunoblot. This experiment was repeated independently twice. c, IC50 of AU-15330 in a panel of human-derived cancer or normal cell lines after 5 days of treatment. Known SMARCA4 loss-of-function (LOF) alterations and multiple myeloma (MM) cell lines with MYC rearrangements (MYC-R’ed) are identified below the graph. AR and FOXA1 scores quantify their transcriptional activities using cognate multi-gene signatures.Source dataUsing a panel of normal and cancer cell lines from 14 distinct lineages, we found AR and FOXA1-driven prostate cancer cells to be preferentially sensitive to AU-15330 (half-maximal inhibitory concentrations (IC50) 1,000 nM) to AU-15330. We observed a similar cytotoxicity profile for ACBI1 and BRM014, an allosteric dual inhibitor of SMARCA2 and SMARCA4 ATPase activity25 (Extended Data Fig. 1h, i). Notably, AR+FOXA1+ prostate cancer cells were more sensitive to these inhibitors than SMARCA4-null cancer cell lines. Several MYC-driven multiple myeloma cells and oestrogen receptor- and/or AR-positive breast cancer cells were also acutely sensitive to AU-15330 (Fig. 1c, Extended Data Fig. 1j, k).In several prostate cancer cell lines, we detected substantial expression of both SWI/SNF ATPases, which were rapidly degraded in a dose-dependent manner by AU-15330 (Extended Data Fig. 2a, b). Concordantly, AU-15330 attenuated the growth of these cells and induced apoptotic cell death, while having no anti-proliferative effect on benign or non-neoplastic prostate cells (grey bars, Fig. 1c) at parallel doses (Extended Data Fig. 1f, 2c–e). Treatment with either the bromodomain ligand alone (AU-15139) or an inactive epimer of AU-15330 (AU-16235) had no effect on target protein levels or cancer cell survival and growth (Extended Data Figs. 1f, g, 2f, g). Next, competition of AU-15330 with a free VHL ligand (VL285), but not with thalidomide, reversed degradation of SWI/SNF targets (Extended Data Fig. 2g) and rescued the growth inhibitory effect in a dose-dependent manner (Extended Data Fig. 2h). Furthermore, pre-treatment of VCaP cells (an AR+FOXA1+ prostate cancer cell line model) with bortezomib (a proteasome inhibitor) or MLN4924 (a NEDD8-activating enzyme inhibitor) hindered target protein degradation, indicating that AU-15330 requires the proteasome machinery and ubiquitination cascade for its action (Extended Data Fig. 2g).As SWI/SNF complexes actively remodel nucleosomal DNA packaging, we profiled the effect of AU-15330 on physical chromatin accessibility using the assay for transposase-accessible chromatin followed by sequencing (ATAC-seq). We detected a rapid and near-complete loss in chromatin accessibility at more than 30,000 sites in VCaP cells with as little as 1 h of AU-15330 treatment (Fig. 2a), which is within minutes of SMARCA2 and SMARCA4 degradation (Extended Data Fig. 3a); approximately 25,000 genomic sites showed little to no change in nucleosomal density (Extended Data Fig. 3b). Similar profound changes in chromatin accessibility were not observed upon treatment with a BRD4 degrader (ZBC-260; Extended Data Fig. 3a, b). In our genetic models using CRISPR–Cas9 and shRNA-mediated target inactivation, we detected a significant compaction of the chromatin only upon concurrent loss of both SWI/SNF ATPases (Extended Data Fig. 3c, d). More than 90% of the AU-15330-compacted sites were within distal regulatory regions, which were enriched for enhancers, whereas the retained sites were predominantly within promoters (Fig. 2b). De novo motif and binding analysis for the regulation of transcription (BART) analyses of AU-15330-compacted sites identified DNA-binding elements for major oncogenic transcription factors in prostate cancer, including AR, FOXA1, HOXB13 and ERG (Extended Data Fig. 3e, f). As expected, retained promoter sites showed enrichment for PolII and E2F motifs (Extended Data Fig. 3g). Interrogation of chromatin changes in LNCaP cells upon AU-15330 treatment reproduced these findings (Extended Data Fig. 4a–c).Fig. 2: SWI/SNF ATPase degradation disrupts physical chromatin accessibility at the core-enhancer circuitry to disable oncogenic transcriptional programs.a, ATAC-seq read-density heat maps from VCaP cells treated with DMSO or AU-15330 for indicated durations (n = 2 biological replicates). b, Genome-wide changes in chromatin accessibility upon AU-15330 treatment for 4 h in VCaP cells along with genomic annotation of sites that lose physical accessibility (lost) or remain unaltered (retained). c, d, ChIP–seq read-density heat maps for AR and FOXA1 (c) and H3K27Ac (d) at the AU-15330 (AU)-compacted genomic sites in VCaP cells after treatment with DMSO or AU-15330 (1 μM) for indicated times and stimulation with R1881 (1 nM, 3 h). e, RNA-seq heat maps for classical AR target genes in LNCaP, VCaP and LAPC4 prostate cancer cells with or without 24 h of AU-15330 treatment.Source dataConcurrent with the loss of accessibility, chromatin immunoprecipitation followed by sequencing (ChIP–seq) revealed a decrease in chromatin binding of AR, FOXA1, and ERG in VCaP cells within 1 h of AU-15330 treatment (Fig. 2c, Extended Data Fig. 4d, e). We also detected disappearance of the characteristic ‘valley’ pattern in the H3K27Ac ChIP–seq signal, indicating the movement of flanking nucleosomes towards the centre of AU-15330-compacted enhancers (Fig. 2d). At early time points, we detected no loss in the abundance of the H3K27Ac mark; however, it was significantly depleted 24 h after AU-15330 treatment (Extended Data Fig. 4f). Similar results were observed upon AU-15330 treatment of LNCaP cells (Extended Data Fig. 4g, h). Loss of AR, FOXA1 and H3K27Ac ChIP signals was evident at enhancer sites of the classical AR target gene KLK3 (Extended Data Fig. 4i). We found AR, FOXA1, ERG and SMARCC1 to co-occupy a large fraction of H3K27Ac-marked regulatory elements (Extended Data Fig. 5a–c). Furthermore, multiple core SWI/SNF components were present in the mass spectrometry-based datasets of AR, FOXA1, and ERG interactomes (Extended Data Fig. 5d), which we confirmed by reciprocal co-immunoprecipitation assays (Extended Data Fig. 5e). This positions SWI/SNF complexes as common chromatin cofactors of the oncogenic transcriptional machinery in prostate cancer cells. As an important control, we saw no changes in chromatin binding of CTCF in AU-15330-treated cells (Extended Data Fig. 6a–d).Global transcriptomic profiling with RNA sequencing (RNA-seq) revealed significant downregulation of AR and FOXA1-regulated genes in multiple prostate cancer cells, as well as ERG-regulated transcripts in ERG fusion-positive VCaP cells. We also detected significant loss in the expression of MYC target genes with AU-15330 (Fig. 2e, Extended Data Fig. 6e, f). The global AU-15330 gene signature was highly concordant with transcriptional changes associated with ARID1A loss (Extended Data Fig. 6g). However, neither BRD7 nor BRD9 degradation alone attenuated the expression of classical AR, FOXA1 and ERG target genes or the MYC gene to an extent comparable to AU-15330, suggesting that canonical SWI/SNF (cBAF) complexes are the primary cofactors of oncogenic enhancer-binding transcription factors (Extended Dat
https://www.nature.com/articles/s41586-021-04246-z
Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer