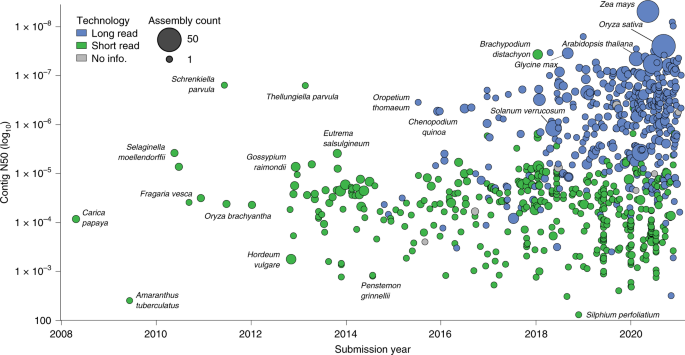
Representation and participation across 20 years of plant genome sequencing The field of plant genome sequencing has grown rapidly in the past 20 years, leading to increases in the quantity and quality of publicly available genomic resources. The growing wealth of genomic data from an increasingly diverse set of taxa provides unprecedented potential to better understand the genome biology and evolution of land plants. Here we provide a contemporary view of land plant genomics, including analyses on assembly quality, taxonomic distribution of sequenced species and national participation. We show that assembly quality has increased dramatically in recent years, that substantial taxonomic gaps exist and that the field has been dominated by affluent nations in the Global North and China, despite a wide geographic distribution of study species. We identify numerous disconnects between the native range of focal species and the national affiliation of the researchers studying them, which we argue are rooted in colonialism—both past and present. Luckily, falling sequencing costs, widening availability of analytical tools and an increasingly connected scientific community provide key opportunities to improve existing assemblies, fill sampling gaps and empower a more global plant genomics community. The pace of sequencing and quality of land plant (Embryophyta) genome assemblies have increased dramatically over the past 20 years. Since the genome assembly of Arabidopsis thaliana—the first for any land plant—was published in 20001, hundreds of plant genomes have been sequenced, assembled and made publicly available on GenBank2 and other repositories for genomic data. With large, complex genomes and varying levels of ploidy, plant genomes have been historically difficult to assemble. However, technological advances, such as long-read sequencing and new computational tools, have made sequencing and assembly of virtually any species possible3,4,5. Here, we provide an overview of the first 20 years of plant genome sequencing, including assessments of assembly quality, taxonomic representation and geographic participation.Land plants are extremely diverse, with publicly available genome assemblies now spanning over ~500 million years of evolution6,7,8. However, only a small fraction (~0.16%) of the ~350,000 extant land plants have had their genome sequenced, and these efforts have not been evenly distributed across clades9. For some plants (for example, maize, Arabidopsis and rice10,11,12), multiple, high-quality genome assemblies are available and thousands of accessions, cultivars and ecotypes have been resequenced using high-coverage Illumina data13. Brassicaceae, a medium-sized plant family (~3,700 species14), is the most heavily sequenced, with genome assemblies for dozens of species including Arabidopsis and numerous cruciferous vegetables. In contrast, for most other groups, none or only a single species has a genome assembly. Ambitious efforts to fill taxonomic sampling gaps exist, including the Earth BioGenome and 10KP projects15,16, but individual research groups also play a role in expanding taxonomic representation in plant genomics.With rapidly expanding resources and a new generation of scientists being trained, now is an ideal time to assess progress in terms of both taxonomic diversity and geographic representation in plant genome science. Economic disparities between nations, many of which were established due to colonialism, have a substantial impact on participation in science. Imperial colonialism provided scientists from the Global North access to a wealth of biodiversity, raw materials and ideas that would have been inaccessible to them otherwise17,18,19. Colonial scientists capitalized on this opportunity and, over time, this led to a disproportionate accumulation of wealth and scientific resources in the Global North20, which has contributed to the establishment and maintenance of global inequality17,19,21. Today, differences in funding, training opportunities, publication styles and language requirements continue to drive similar inequities19,22,23,24. In genomics, the high costs of sequencing and computational resources are barriers to entry that perpetuate existing imbalances established due to colonialism and economic disparities. Luckily, the diminishing cost and increasing accessibility of sequencing and computation infrastructure provide an opportunity to broaden participation and increase equity in genomics. This will require affluent nations and individuals to recognize their disproportionate access to biological and genetic resources and seek to increase participation rather than capitalizing on their privilege.Here, we provide a high-level perspective on the first 20 years of genome sequencing in land plants. We describe the taxonomic distribution of sequencing efforts and build on previous estimates of genome availability and quality25,26,27,28. We show that an impressive and growing number of land plant genome assemblies are now publicly available, that quality has greatly improved in concert with the rise of long-read sequencing but that substantial taxonomic gaps exist. We also describe the geographic landscape of plant genomics, with an emphasis on representation. We highlight the need for the field, including its many affluent researchers and institutions, to work towards broadening participation. In our view, the wealth of publicly available genome assemblies can be leveraged to better understand plant biology while also continuing to decolonize a major field of research.As of January 2021, 798 land plant species have genome assemblies. Six hundred and thirty-one of these were deposited in GenBank, and we identified a further 167 with genome assemblies via literature searches and cross-referencing against additional databases. If multiple genome assemblies were available for a species, we selected the highest-quality genome assembly (based on contiguity) as a representative for that species. Unless otherwise noted, all analyses were conducted on this dataset of 798 genome assemblies (Supplementary Table 1).The quantity and quality of land plant genome assemblies have increased rapidly, with particularly notable improvements associated with the advent of long-read sequencing (Fig. 1). Overall, 74% of land plant genome assemblies were produced in the past 3 years. Contig N50 (the length of the shortest contig in the set of contigs containing at least 50% of the assembly length) has also increased markedly in recent years, from 99.5 ± 48.1 kb in 2010 to 3,395.2 ± 735.4 kb in 2020. This increase appears to be driven primarily by advances in sequencing technologies. Assemblies constructed with short-read technology (for example, Illumina and Sanger) have significantly lower (P < 0.0001) contig N50 (124.6 ± 58.2 kb) compared to those that incorporate long reads (for example, PacBio and Oxford Nanopore) with a contig N50 of 4,033.4 ± 618.9 kb. This difference translates to an impressive ~32-fold increase in mean contig N50 for long-read assemblies. Nevertheless, many extremely fragmented plant genome assemblies have been published. Twenty-three of the assemblies in our dataset have a contig N50 <1 kb, and 158 with <10 kb.Fig. 1: Changes in land plant genome assembly quality and availability over time.Assembly contiguity by submission date for 798 land plant species with publicly available genome assemblies. Points are coloured by the type of sequencing technology used and scaled by the number of assemblies available for that species. There is an improvement in contiguity associated with the advent of long-read sequencing technology, and a noticeable increase in the number of genome assemblies generated annually. All assemblies generated before 2008 have since been updated and are therefore not included.The first land plants to have their genomes sequenced and assembled were model or crop species with small diploid genomes, but it is now feasible to assemble a genome for virtually any taxon. Nevertheless, taxonomic sampling gaps persist. Of the 137 land plant orders that have been described29, over half (76) lack a representative genome assembly. For the 62 orders with at least one genome assembly, a wide range of sampling depth is evident. For example, there are 83 species with genome assemblies in Brassicales, 80 in Poales and 67 in Lamiales, yet there are 41 orders with ten or fewer sequenced species. Six orders of land plants are statistically over-represented in genome assembly databases based on species richness. These include the agriculturally and economically important clades of Brassicales, Cucurbitales, Fagales, Malvales, Rosales and Solanales. Four orders of land plants had significantly fewer genome assemblies than expected based on species richness (Fig. 2). Not surprisingly, these were speciose orders with notable ecological but comparatively less economic importance—Asparagales, Asterales, Gentianales and Polypodiales (Fig. 2a). Bryophytes are poorly represented, with assemblies for only eight mosses, three liverworts and three hornworts (Fig. 2a and Extended Data Fig. 1). Diploid species are also statistically over-represented in terms of genome assembly availability (Fig. 2b and Extended Data Fig. 2) despite the widespread occurrence of polyploid plants30. Until recently, technological limitations have made it difficult to assemble high-quality polyploid genomes4. However, with the improvements offered by long-read sequencing, it is becoming more feasible to sequence and assemble large-polyploid plant genomes. As a result, there are some highly contiguous tetraploid and reasonably contiguous hexaploid genome assemblies, with mean contig N50 of 1,855.7 ± 474.3 and 251.9 ± 99.8 kb, respectively (Extended Data Fig. 2).Fig. 2: Comparison of genome availability and quality metrics for each land plant order.a, The number of species with publicly available genome assemblies as of January 2021 (n = 798) versus the number expected for each order. Significance values were calculated using Fisher's exact test. Orders with no genome assemblies are shown in grey. Bryophytes are plotted at the phylum level, but Extended Data Fig. 2 shows bryophyte orders. Orders showing significant over- or under-representation are marked with asterisks. Over-represented orders include Brassicales (P = 3.03 × 10–13), Cucurbitales (P = 0.0038), Fagales (P = 0.0003), Malvales (P = 0.0084), Rosales (P = 0.0286) and Solanales (P = 1.27 × 10–6). Under-represented orders include Asparagales (P = 2.62 × 10–11), Asterales (P = 1.00 × 10–10), Gentianales (P = 0001) and Polypodiales (P = 8.93 × 10–8). b, Box plots showing the distribution of assembly length for each order of land plants. Points are coloured by ploidy. c, Box plots showing the distribution of contig N50 for each order of land plants. d, Box plots showing the distribution of complete BUSCO percentages for each order of land plants. c,d, Points are coloured by sequencing technology. For all box plots, the box defines the interquartile range (25th–75th percentile) and the centre line represents the median; whiskers extend to the maximum and minimum data values.To further assess differences in assembly quality and completeness, we quantified the percentage of benchmarking universal single-copy orthologues (BUSCO, v.4.1.421) using the Embryophyta gene set from OrthoDB v.10 (ref. 31) that were present in each land plant genome assembly deposited in GenBank. There was a high degree of variability in BUSCO completeness: percentages of complete BUSCOs (single and duplicated genes) ranged from 0% to 99% across the available genome assemblies (Fig. 2d). More contiguous genome assemblies with higher contig N50 had more complete BUSCOs (P < 0.0088), and this was associated with the use of long reads in the assembly process (P < 0.0001; Fig. 2c–d and Extended Data Fig. 3). Despite the wide range of BUSCO completeness, no significant associations between the percentage of BUSCOs and genome size, taxonomy or domestication status were identified.To quantify whether a bias exists towards sequencing economically important plants relative to other species, we classified the domestication status of each species with a genome assembly into six categories: (1) domesticated: plants that have undergone extensive artificial selection; (2) cultivated: plants that are used by humans but have not been subjected to substantial artificial selection; (3) natural commodity: plants that are harvested with little cultivation; (4) feral: plants that are not economically important but have still been influenced by human selection; (5) wild: plants that occur in the wild and have not been directly impacted by humans; and (6) wild relatives: wild plants that are closely related to or progenitors of domesticated and cultivated crops. Based on these categories, genome assemblies are available for 135 domesticated, 127 cultivated, 120 natural commodity and 12 feral species. The remaining 404 genome assemblies are from wild species; of these, 77 are wild relatives of crops (Fig. 3). While the number of human-linked species (that is, domesticated, cultivated, natural commodity and feral) with genome assemblies is largely equivalent to wild species, this equivalence reflects an extreme bias. There are far more wild (~350,000)32 than domesticated species (~1,200–2,000)33,34, suggesting that wild plants represent an untapped reservoir of genomic information.Fig. 3: Geographic distribution of the submitting institutions for 798 plant genome assemblies.Circles are scaled by the number of genome assemblies produced in each nation and coloured by the relative proportion of domesticated, cultivated, feral, natural commodity, wild and wild relative species sequenced.To better understand global participation in plant genomics, we identified the submitting institution for each genome assembly in our dataset. If the submitting institution was not listed, we identified the corresponding author for the associated publication and assigned the genome to the location of that institution. While this approach does not account for secondary affiliations in other nations, it does reveal where most of the scientific credit for a genome assembly is probably placed.
https://www.nature.com/articles/s41477-021-01031-8
Representation and participation across 20 years of plant genome sequencing