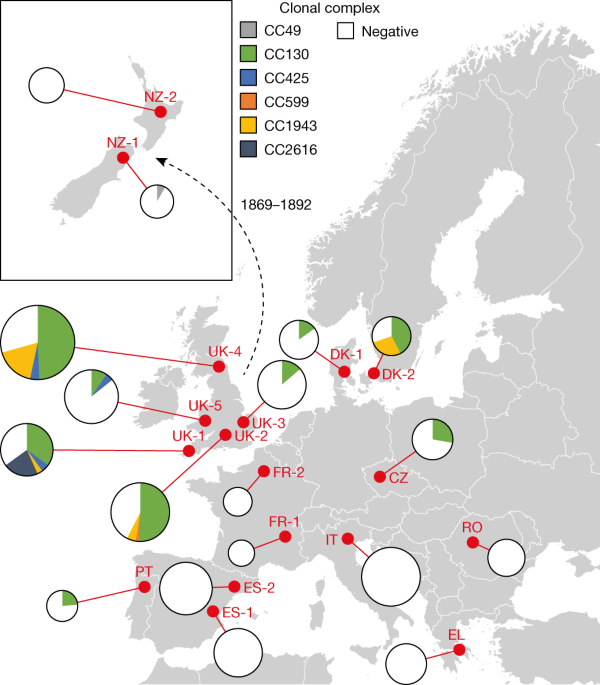
Emergence of methicillin resistance predates the clinical use of antibiotics Jesper Larsen orcid.org/0000-0003-0582-04571 na1, Claire L. Raisen2 na1, Xiaoliang Ba orcid.org/0000-0002-3882-35852, Nicholas J. Sadgrove3, Guillermo F. Padilla-González orcid.org/0000-0002-8300-68913, Monique S. J. Simmonds3, Igor Loncaric4, Heidrun Kerschner5, Petra Apfalter5, Rainer Hartl5, Ariane Deplano6, Stien Vandendriessche6 nAff46, Barbora Černá Bolfíková orcid.org/0000-0001-8059-48897, Pavel Hulva8,9, Maiken C. Arendrup1, Rasmus K. Hare1, Céline Barnadas1,10, Marc Stegger1, Raphael N. Sieber1, Robert L. Skov orcid.org/0000-0002-6079-538111, Andreas Petersen1, Øystein Angen1, Sophie L. Rasmussen orcid.org/0000-0002-2975-678X12,13, Carmen Espinosa-Gongora orcid.org/0000-0002-9536-054814, Frank M. Aarestrup orcid.org/0000-0002-7116-272315, Laura J. Lindholm16, Suvi M. Nykäsenoja17, Frederic Laurent18, Karsten Becker orcid.org/0000-0002-6391-134119, Birgit Walther20 nAff47, Corinna Kehrenberg21, Christiane Cuny22, Franziska Layer orcid.org/0000-0002-4613-647822, Guido Werner22, Wolfgang Witte22, Ivonne Stamm23, Paolo Moroni orcid.org/0000-0002-0974-308424 nAff48, Hannah J. Jørgensen orcid.org/0000-0002-1788-921925, Hermínia de Lencastre orcid.org/0000-0001-6816-893226,27, Emilia Cercenado orcid.org/0000-0002-5279-377328, Fernando García-Garrote28 nAff49, Stefan Börjesson orcid.org/0000-0003-2219-265929 nAff50, Sara Hæggman30, Vincent Perreten31, Christopher J. Teale32, Andrew S. Waller orcid.org/0000-0002-7111-954933 nAff51 nAff52, Bruno Pichon34, Martin D. Curran35, Matthew J. Ellington35 nAff53, John J. Welch36, Sharon J. Peacock orcid.org/0000-0002-1718-278237, David J. Seilly2, Fiona J. E. Morgan2 nAff54, Julian Parkhill orcid.org/0000-0002-7069-59582, Nazreen F. Hadjirin2, Jodi A. Lindsay38, Matthew T. G. Holden orcid.org/0000-0002-4958-216639, Giles F. Edwards40, Geoffrey Foster orcid.org/0000-0002-5527-758X41, Gavin K. Paterson orcid.org/0000-0002-1880-009542, Xavier Didelot orcid.org/0000-0003-1885-500X43, Mark A. Holmes orcid.org/0000-0002-5454-16252 na2, Ewan M. Harrison orcid.org/0000-0003-2720-050737,44,45 na2 & Anders R. Larsen1 na2 Nature (2022)Cite this article The discovery of antibiotics more than 80 years ago has led to considerable improvements in human and animal health. Although antibiotic resistance in environmental bacteria is ancient, resistance in human pathogens is thought to be a modern phenomenon that is driven by the clinical use of antibiotics1. Here we show that particular lineages of methicillin-resistant Staphylococcus aureus—a notorious human pathogen—appeared in European hedgehogs in the pre-antibiotic era. Subsequently, these lineages spread within the local hedgehog populations and between hedgehogs and secondary hosts, including livestock and humans. We also demonstrate that the hedgehog dermatophyte Trichophyton erinacei produces two β-lactam antibiotics that provide a natural selective environment in which methicillin-resistant S. aureus isolates have an advantage over susceptible isolates. Together, these results suggest that methicillin resistance emerged in the pre-antibiotic era as a co-evolutionary adaptation of S. aureus to the colonization of dermatophyte-infected hedgehogs. The evolution of clinically relevant antibiotic-resistance genes in wild animals and the connectivity of natural, agricultural and human ecosystems demonstrate that the use of a One Health approach is critical for our understanding and management of antibiotic resistance, which is one of the biggest threats to global health, food security and development. Methicillin-resistant S. aureus (MRSA) is one of the most common antibiotic-resistant bacterial pathogens, causing approximately 171,000 invasive infections each year in Europe alone2. MRSA was first identified in 1960 shortly after the introduction of methicillin (celbenin) as a treatment option against penicillin-resistant S. aureus clones3, but was possibly selected for by the clinical use of penicillin over the previous 20 years4. Methicillin resistance has subsequently emerged in many S. aureus clones around the world, both in hospital and community settings as well as in livestock such as pigs and cattle5,6. This has serious implications for the treatment of severe infections and the World Health Organization now considers MRSA to be an important threat to human health7.Methicillin resistance in S. aureus is mediated by the mecA and mecC genes, which encode the enzymes penicillin-binding protein 2a (PBP2a) and PBP2c, respectively. mecA and mecC confer resistance to almost all β-lactam antibiotics, including penicillinase-labile penicillins (such as penicillin G), penicillinase-stable penicillins (such as methicillin) and cephalosporins (such as cefoxitin).Hedgehog surveys from Denmark and Sweden demonstrated a surprisingly high prevalence of MRSA carrying mecC (mecC-MRSA)8,9, raising the possibility that the evolution of these bacteria was driven by natural selection in wildlife, as opposed to clinical use of antibiotics. Historically, mecC-MRSA was first discovered in dairy cows and subsequently in humans10, suggesting that the use of antibiotics in livestock was providing a selective advantage and that human infections were the result of zoonotic transmission. Studies from many different European countries revealed that mecC-MRSA is also present in other domesticated animals such as sheep, goats and horses as well as in a broad range of wild animals, albeit at low frequencies11.Our hypothesis that the evolution of mecC-MRSA was driven by natural selection is supported by studies from northwestern Europe and New Zealand that showed that hedgehogs are frequently colonized with the dermatophyte T. erinacei, which produces a penicillinase-labile penicillin-like substance that was recently identified as penicillin G12,13,14,15,16,17,18,19. To test our hypothesis, we examined the distribution of mecC-MRSA and other S. aureus isolates in hedgehogs in ten European countries and New Zealand. We sequenced 244 S. aureus isolates from hedgehogs and 913 S. aureus isolates from other sources to infer the evolutionary histories, host dynamics, geographical dispersal patterns and zoonotic potential of the major mecC-MRSA clones in Europe. The potential mechanisms for the natural selection of mecC-MRSA by T. erinacei were assessed by analysing the genome of T. erinacei for β-lactam biosynthetic genes and by screening T. erinacei for the production of β-lactams and antibiotic activity against a panel of S. aureus strains.We first examined the geographical distribution and population structure of mecC-MRSA in European hedgehogs, which inhabit large parts of Europe as a result of postglacial expansion from Pleistocene refugia20. European hedgehogs have also become widespread in New Zealand after a series of introductions from the UK between 1869 and 1892 (ref. 21). We analysed 828 samples from the nasal area, skin and feet of 276 hedgehogs originating from 16 wildlife rescue centres in 10 European countries and 2 wildlife rescue centres in New Zealand (Fig. 1 and Extended Data Fig. 1). mecC-MRSA was present in 101 of the 172 hedgehogs (222 out of 516 samples) from England and Wales (66%, 81 out of 123), Czech Republic (50%, 6 out of 12), Denmark (50%, 11 out of 22), Portugal (29%, 2 out of 7) and New Zealand (6%, 1 out of 17), therefore extending the known geographical distribution of mecC-MRSA in hedgehogs (Fig. 1 and Extended Data Fig. 1). By contrast, all 104 hedgehogs (312 samples) from Greece, Romania, Italy, France and Spain tested negative for mecC-MRSA. Whole-genome sequencing showed that the 222 mecC-MRSA isolates belonged to 6 clonal complexes, CC130 (75%), CC1943 (15%), CC2616 (6%), CC425 (3%), CC49 (1%) and CC599 (1%), of which CC130 had the most widespread distribution across western and central Europe (Fig. 1 and Extended Data Fig. 1). We screened all of the MRSA-negative hedgehog samples from our study (n = 606) for the presence of methicillin-susceptible S. aureus (MSSA) isolates belonging to the same clonal complexes as the mecC-MRSA isolates (Extended Data Fig. 1). This led to the identification of 22 MSSA isolates, including 13 CC49 isolates from Spain (n = 9), Denmark (n = 3) and Portugal (n = 1), and 9 CC130 isolates from England (n = 8) and Spain (n = 1).Fig. 1: Distribution of mecC-MRSA clones in European and New Zealand hedgehog samples.The analysis included 828 samples from the nasal area, skin and feet of 276 hedgehogs originating from 16 wildlife rescue centres in 10 European countries and 2 wildlife rescue centres in New Zealand. The red dots indicate the sampling locations. The pie charts are connected to the sampling locations by a red line. The area of the pie chart is proportional to the number of samples from that location. The introduction of European hedgehogs into New Zealand from the UK between 1869 and 1892 is shown. A detailed description of the results is provided in Extended Data Fig. 1. Maps were provided by Eurostat under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence; the administrative boundaries are copyright of EuroGeographics.Source dataThe mecC gene encoding PBP2c is located immediately upstream of a blaZ gene (hereafter, blaZLGA251) on a chromosomally integrated mobile genetic element known as a type XI staphylococcal cassette chromosome mec (SCCmec). PBP2c and the blaZLGA251-encoded penicillinase are orthologues of the PBP2a enzyme and penicillinase produced by other S. aureus clones, although they share only 63% and 65% amino acid identities with each other, respectively10. Penicillinases have a narrower spectrum than PBP2a and PBP2c and provide resistance only to penicillin G and other penicillinase-labile subclasses of penicillin. As expected, blaZLGA251 was present in the 222 mecC-MRSA isolates but absent in the 22 MSSA isolates. However, 14 of the MSSA isolates carried the blaZ gene found in other S. aureus clones (Supplementary Table 1).The abundance of mecC-MRSA in hedgehogs led us to speculate that antibiotic production by T. erinacei provides a selective environment in which mecC-MRSA isolates have an advantage over susceptible isolates. Genome sequencing and analysis of the T. erinacei type strain IMI 101051 (ATCC 28443) identified orthologues of pcbAB, pcbC and penDE, which are responsible for key steps in penicillin G production by Penicillium chrysogenum, as well as the Acremonium chrysogenum early cephalosporin C biosynthetic genes cefD1 and cefD2, which are involved in the conversion of isopenicillin N into penicillin N (Fig. 2 and Extended Data Table 1). By contrast, T. erinacei IMI 101051 lacked the A. chrysogenum late cephalosporin C biosynthetic genes cefEF and cefG. P. chrysogenum also carries cefD1 and cefD2 but is nevertheless incapable of producing cephalosporins due to the lack of cefEF and cefG22,23.Fig. 2: Penicillin biosynthetic genes and antibiotic activity of T. erinacei IMI 101051.a, Schematic of the key steps in the biosynthesis of penicillin G and cephalosporin C. The presence (green) or absence (red) of T. erinacei penicillin G and cephalosporin C biosynthetic genes is indicated. b, T. erinacei inhibition zones against a collection of S. aureus control strains (black) and two mecC-MRSA wild-type strains belonging to CC130 (green) and CC425 (blue) and their isogenic mutants. Two-tailed paired Student’s t-tests were used to compare inhibition zones of each mutant to the corresponding wild-type strain. Data are mean ± s.d.; n = 4 biologically independent fungal culture extracts. A detailed description of the results is provided in Extended Data Fig. 4.Source dataWe processed four distinct culture broths of T. erinacei IMI 101051 for metabolic profiling using liquid chromatography–mass spectrometry (LC–MS) and molecular networking analysis. This led to the identification of two β-lactams, penicillin G and 6-(5-hydroxy-n-valeramido)-penicillanic acid (KPN), both of which belong to the penicillin class of antibiotics (Extended Data Figs. 2 and 3 and Supplementary Fig. 1). KPN has to date been found only in culture broths of fungal strains belonging to the genus Paecilomyces24 and differs from penicillin G by having a unique side chain (Extended Data Figs. 2 and 3 and Supplementary Fig. 1). The biosynthetic pathway of KPN is currently unknown.Four culture broths of T. erinacei IMI 101051 were screened for antibiotic activity against a collection of S. aureus control strains. All of the culture broths produced large inhibition zones against two penicillin-susceptible S. aureus strains—ATCC 9144 (Oxford S. aureus) and ATCC 25923—but much smaller zones against the penicillinase-producing S. aureus strain ATCC 29213 and the mecA-positive S. aureus strain ATCC 43300 (Fig. 2 and Extended Data Fig. 4). The role of mecC and blaZLGA251 was assessed by screening the culture broths for antibiotic activity against two mecC-MRSA wild-type strains belonging to CC130 (02.5099.D) and CC425 (LGA251) and their isogenic mutants. The mutants with deleted mecC (ΔmecC), blaZLGA251 (ΔblaZLGA251), and mecC and blaZLGA251 (ΔmecC-blaZLGA251) produced significantly larger inhibition zones compared with the corresponding wild-type strains, although the zones of the ΔblaZLGA251 and ΔmecC-blaZLGA251 mutants were larger compared with the zones of the ΔmecC mutants (Fig. 2 and Extended Data Fig. 4). These results indicate that mecC and blaZLGA251 both contribute to the reduced susceptibility of mecC-MRSA to penicillin G and KPN present in culture broths of T. erinacei IMI 101051.We sought to infer the evolutionary histories of S. aureus CC130, CC425 and CC1943, which constitute the most successful mecC-MRSA clones in Europe10,11,25. For this purpose, we collected and sequenced 786 mecC-MRSA and 127 MSSA CC130, CC425 and CC1943 isolates selected to represent the known geographical distribution (mainly western and central Europe) and host repertoire (mainly humans, cattle, sheep, goats and wild animals) of each clone (Supplementary Table 1). We used core-genome single-nucleotide polymorphism (SNP) diversity and isolation dates to infer time-scaled phylogenies of these isolates and the 205 mecC-MRSA and 9 MSSA CC130, CC425 and CC1943 isolates collected from hedgehogs (Supplementary Table 1). The sequencing data were processed for pan-genome analysis to identify antibiotic-resistance genes (ARGs) and mobile genetic elements that encode human- and ruminant-specific immune modulators that are involved in host switching events, including a phage-encoded immune evasion cluster-1 (IEC-1) enabling S. aureus to evade the human innate immune response and a staphylococcal pathogenicity island (SaPI)-encoded vwb gene (vwbSaPI), which encodes a von Willebrand factor-binding protein with coagulase activity against ruminant plasma26.We also sought to infer a time-scaled phylogeny of the 991 type XI SCCmec elements containing the mecC and blaZLGA251 genes but the correlation between root-to-tip distances and isolation dates was too weak with a coefficient of determination R2 = −0.05 (Extended Data Fig. 5). Instead, we used the topology of the type XI SCCmec phylogeny to identify monophyletic mecC-MRSA lineages harbouring orthologous type XI SCCmec elements. The type XI SCCmec elements could be traced back to seven nodes that were connected to each other on a long backbone. Each of the backbone nodes and its orthologous descendants received the same letter designation to reflect their genetic relationship (A to G) (Fig. 3 and Supplementary Fig. 2). Manual mapping of the tips onto the CC130, CC425 and CC1943 phylogenies, and vice versa, enabled us to assign the mecC-MRSA isolates to 16 monophyletic lineages harbouring orthologous type XI SCCmec elements (Fig. 3 and Supplementary Figs. 2–5). The 129 mecC-MRSA CC1943 isolates could be divided into three lineages (C1 to C3), which probably originated in the early-to-late 1800s, long before the first β-lactam—penic
https://www.nature.com/articles/s41586-021-04265-w
Emergence of methicillin resistance predates the clinical use of antibiotics