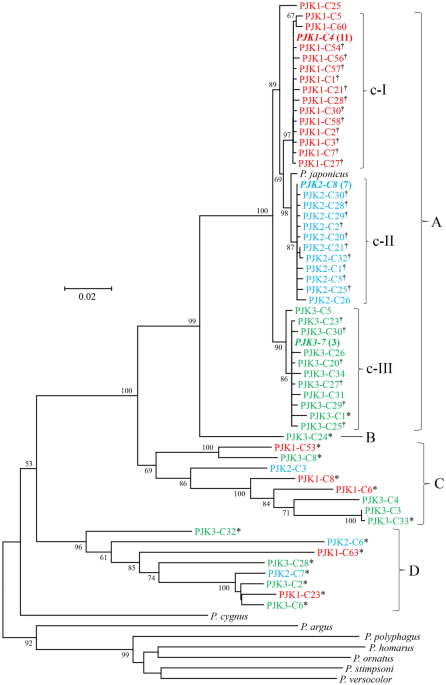
Detection of heteroplasmy and nuclear mitochondrial pseudogenes in the Japanese spiny lobster Panulirus japonicus Partial mtDNA cytochrome oxidase subunit I (COI) fragments and near entire stretch of 12S rDNA (12S) and control region (Dloop) of the Japanese spiny lobster (Panulirus japonicus) (n = 3) were amplified by PCR and used for direct nucleotide sequencing and for clone library-based nucleotide sequence analysis. Nucleotide sequences of a total of 75 clones in COI, 77 in 12S and 92 in Dloop were determined. Haplotypes of the clones matched with those obtained by direct sequencing were determined to be genuine mtDNA sequence of the individual. Phylogenetic analysis revealed several distinct groups of haplotypes in all three regions. Genuine mtDNA sequences were observed to form a group with their closely related variables, and most of these variables may be due to amplification error but a few to be heteroplasmy. Haplotypes determined as nuclear mitochondrial pseudogenes (NUMTs) formed distinct groups. Nucleotide sequence divergence (K2P distance) between genuine haplotypes and NUMTs were substantial (7.169–23.880% for COI, 1.336–23.434% for 12S, and 7.897–71.862% for Dloop). These values were comparable to or smaller than those between species of the genus Panulirus, indicating that integration of mtDNA into the nuclear genome is a continuous and dynamic process throughout pre- and post-speciation events. Double peaks in electropherograms obtained by direct nucleotide sequencing were attributed to common nucleotides shared by multiple NUMTs. Information on the heteroplasmy and NUMTs would be very important for addressing their impact on direct nucleotide sequencing and for quality control of nucleotide sequences obtained. Mitochondrial DNA (mtDNA) has been widely used in molecular phylogenetics, population genetics, and DNA barcoding in animals due to its rapid evolutionary rate, little recombination, strict maternal inheritance, and homoplasmy within individual. On the other hand, there have been increasing number of reports on nuclear mitochondrial pseudogene referred to as ‘NUMT’1,2 and heteroplasmy3,4 in a wide range of eukaryotes. Direct nucleotide sequencing for PCR amplicons of mtDNA has been a conventional tool to detect sequence variation within and between species, however, electropherograms with double peaks or that are completely unreadable are sometimes encountered. These may be attributed to contamination of non-specific amplicons or to point mutations and length variation in heteroplasmic copies or NUMTs. Since stronger signals of the double peaks in electropherogram are preferentially adopted and unreadable electropherograms are discarded as ‘sequencing failed’, the incidences of heteroplasmy and NUMTs are likely to be underestimated. Since the number of heteroplasmic copies and NUMTs may be much smaller than that of the genuine mtDNA molecules, one might expect that heteroplasmy and NUMTs have little negative impact on the quality of nucleotide sequences obtained by direct nucleotide sequencing. However, consistent problems with obtaining good electropherograms in PCR-amplified mtDNA cytochrome oxidase subunit I (COI) fragments have been reported in some crustacean species5,6,7, in which multiple DNA sequences similar to a COI gene were detected from a single individual. In addition to these technical issues, unnoticed incorporation of heteroplasmic copies and NUMTs may lead to overestimation of population diversity and the number of species2,5,8.We have frequently had difficulty for obtaining good electropherograms produced by direct nucleotide sequencing for COI, 12S rDNA (12S) and control region (Dloop) of the Japanese spiny lobster (Panulirus japonicus) and hypothesized that heteroplasmy was the major suspect for the issue. Then, we first begun to investigate extent of heteroplasmy in these regions but eventually found a fair number of NUMTs. To the best of our knowledge, this is the first investigation for the extent of heteroplasmy and NUMTs in the Japanese spiny lobster as well as their impact for direct nucleotide sequencing.Readable electropherograms were obtained from both direction in COI fragments of all three individuals of the Japanese spiny lobster. COI sequences determined by direct nucleotide sequencing ranged from 807 to 864 bp and have been deposited in International Nucleotide Sequence Database Collection (INSDC) under accession numbers of LC571524‒LC571526. No stop codon was observed in these sequences (designated by PJK1-direct, PJK2-direct, and PJK3-direct). No indel was observed between these sequences. All nucleotide substitutions at 19 variable sites observed between these sequences were transition at the 3rd position of a codon, and all substitutions were synonymous. The mean Kimura two parameter (K2P) distance between these three haplotypes was 1.510 ± 0.352% SE and that between these sequences and a reference sequence of P. japonicus (NC_004251) was 1.087 ± 0.270%, which were all well within the range reported for Japanese spiny lobster samples collected in Japan and Taiwan9,10.Electropherograms obtained by forward primer for 12S fragments were not readable, while those by reverse primer were readable in all individuals. 12S sequences determined by direct nucleotide sequencing using reverse primer alone ranged from 551 to 570 bp and have been deposited in INSDC under accession numbers of LC605705‒LC605707. Of nine variable sites, eight were transition and one was indel. The mean K2P distance between these three haplotypes (designated by PJK1-12Sdirect, PJK2-12Sdirect, and PJK3-12Sdirect) was 0.970 ± 0.338%, and that between these sequences and a reference sequence of P. japonicus was 0.835 ± 0.282%.Electropherograms obtained by both primers for Dloop fragments were readable only in one individual (PJK2). This Dloop sequence determined by direct nucleotide sequencing was 762 bp and deposited in INSDC under accession number of LC605749. K2P distance between this haplotype (designated by PJK2-Dloopdirect) and a reference sequence of P. japonicus was 3.666%. No indel was observed between the two sequences, and 25 of 27 variable sites were transition.Phylogenetic analysis of clones, heteroplasmy and NUMTsAmong the 36–42 positive COI clones examined per individual, sequences (809–892 bp) of 22–31 clones per individual (75 clones in total) were successfully determined. After alignment, both ends of all sequences were trimmed to fit the shortest sequence obtained by direct nucleotide sequencing, yielding 774–810 bp sequences. Eleven clones of PJK1 were identical to PJK1-direct, as well as seven of PJK2 to PJK2-direct and three of PJK3 to PJK3-direct. These dominant haplotypes (807 bp) were determined to be genuine COI haplotypes of each individual, and representative sequences of these three genuine haplotypes were deposited in INSDC (LC 571527, LC571533 and LC571538). Nucleotide sequences of the remaining 54 clones were all different one another, in which 20 haplotypes were observed in PJK1, 14 in PJK2, and 20 in PJK3 (LC571541–LC571577, OK429332–OK429343, LC654683-LC654687).Phylogenetic tree constructed using three genuine COI haplotypes, 57 unique haplotypes and eight sequences of reference lobster species is shown in Fig. 1. Haplotypes detected from P. japonicus were segregated into four groups (designated by A, B, C and D). Among the outgroup species used, Australian rock lobster (P. cygnus) that morphologically and genetically belongs to the P. japonicus group11,12, appeared to be the closest kin to all haplotypes detected from P. japonicus. All haplotypes in group A were of the same length (807 bp), and no indel was observed. Three distinct clades (designated by c-I to c-III) were observed in group A, in which 14 haplotypes from PJK1, 11 from PJK2 and 11 from PJK3 were cohesively clustered together with their corresponding genuine haplotypes (bold italic). PJK1-C25 was outlier, having 10 nucleotide differences from the genuine COI sequence. The numbers of variable nucleotide sites between haplotypes within c-I, c-II and c-III were 20, 15 and 26, respectively, of which nonsynonymous nucleotide substitutions were observed at 11, 13 and 10 sites. Stop codon was observed only in one haplotype (PJK3-C1). The mean K2P distance between different haplotypes within these clades ranged from 0.320 ± 0.075 to 0.561 ± 0.103%. The mean K2P distances between three clades ranged from 1.343 ± 0.339 to 2.178 ± 0.464%. Although group A must be composed of sequences containing those caused by Taq polymerase error or true heteroplasmic sequences as well as genuine haplotypes, it is difficult to determine the former two categories. All of the non-genuine haplotypes in group A had singleton difference one another, supporting the occurrence of Taq polymerase error. We determined haplotypes (marked with dagger in Fig. 1) differed by less than two substitutions from the genuine haplotype to be due to Taq polymerase error. This criterion may be reasonable, since Taq polymerase-mediated errors were estimated to occur approximately at a frequency of 7.2 × 10−5 per bp per cycle13 to one mutation per 10,000 nucleotides per cycle14. When Taq polymerase error is taken into account, these K2P distances within and between clades and number of haplotypes are likely to be somewhat overestimated. PJK1-C25, two (PJK1-C5 and PJK1-C60) in c-I clade, one (PJK2-C26) in c-II, and five (PJK3-C1, PJK3-C5, PJK3-C26, PJK3-C31, PJK3-C34) in c-III differed by 3 to 10 nucleotides from their genuine haplotypes, which were determined to be heteroplasmic haplotypes.Figure 1Neighbor-joining phylogenetic (NJ) tree showing relationships among 57 different haplotypes of cytochrome oxidase subunit I (COI) or COI-like sequences obtained from the Japanese spiny lobster (Panulirus japonicus), and COI sequences of eight congeneric species derived from the GenBank database. Haplotypes detected from the same individual of the Japanese spiny lobster share the same color. Genuine mtDNA haplotype is shown in bold italic and number of clones examined is shown in parenthesis. Stop codons were observed in haplotypes carrying asterisk. Haplotypes carrying dagger differ from the corresponding genuine mtDNA haplotype by less than two nucleotides (including indel). The bootstrap values greater than 60% (out of 1000 replicates) are shown at the nodes.Sequence size of haplotypes in groups B to D ranged from 774 to 810 bp. K2P distance between haplotypes of groups A and B ranged from 7.169 to 8.177% with a mean of 7.754 ± 0.973%, that between A and C ranged from 12.073 to 17.392% with a mean of 14.521 ± 1.151%, and that between A and D ranged from 17.472 to 23.880% with a mean of 21.042 ± 1.600%. Multiple stop codons were observed in a haplotype of group B, in five of eight haplotypes of group C, and all haplotypes of group D. Three haplotypes in group C had no stop codon but differed in four to 10 deduced amino acids from the genuine haplotypes. BLAST homology search revealed no identical sequence for haplotypes in groups B to D but indicated that the closest species were P. japonicus or P. cygnus with moderate similarity (83–89% homology). Therefore, all haplotypes of groups B to D (LC571565–LC571570, LC571572–LC571577, LC654683-LC654687) were determined to be NUMTs.Among the 30–35 positive 12S clones examined per individual, sequences (772–806 bp) of 25–27 clones per individual (77 clones in total) were successfully determined. After alignment, primer sequences were trimmed, yielding 731–765 bp sequences. Thirteen clones of PJK1 were identical one another, as well as 12 of PJK2 and three of PJK3, and these were identical to PJK1-12Sdirect, PJK2-12Sdirect and PJK3-12Sdirect, respectively. These dominant haplotypes ranging from 761 to 762 bp in size were determined to be genuine 12S haplotypes of the individual, and representative sequences of these three genuine haplotypes were deposited in INSDC (LC605708‒LC605710). Nucleotide sequences of the remaining 49 clones were all different one another, in which 12 haplotypes were observed in PJK1, 23 in PJK2, and 14 in PJK3 (LC605711‒LC605748, OK429126–OK429131, LC654678-LC654682).Since incorporation of all eight Panulirus species sequences made sequence alignment ambiguous because of multiple indels, reference sequences of P. japonicus and of closely related P. cygnus were used for constructing phylogenetic tree (Fig. 2). Haplotypes detected from P. japonicus were segregated into three groups (designated by A to C). Sequence size of haplotypes in group A ranged from 760 to 762 bp. Three distinct clades (s-I to s-III) were observed in group A, in which 12 haplotypes each from PJK1, PJK2 and PJK3 were cohesively clustered together with their corresponding genuine haplotypes (bold italic). The numbers of variable nucleotide sites between haplotypes within s-I, s-II and s-III were 24, 17 and 16, respectively. Of these variable sites, transversion was observed at five, one and three sites, and indel was observed at one, zero and one sites, respectively. The mean K2P distances between different haplotypes within these clades ranged from 0.345 ± 0.081 to 0.519 ± 0.101%. The mean K2P distances between three clades ranged from 0.936 ± 0.275 to 1.371 ± 0.359%. Haplotypes differed by less than two substitutions (including indel) from
https://www.nature.com/articles/s41598-021-01346-8
Detection of heteroplasmy and nuclear mitochondrial pseudogenes in the Japanese spiny lobster Panulirus japonicus