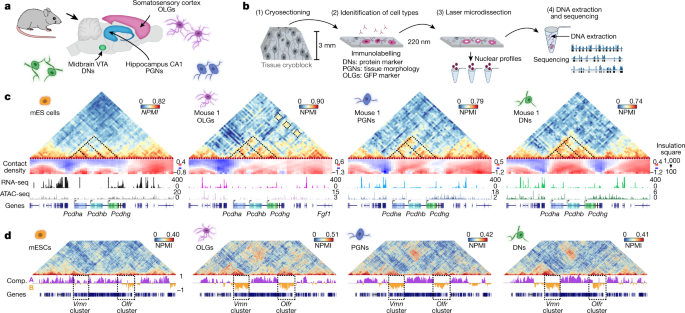
Cell-type specialization is encoded by specific chromatin topologies Warren Winick-Ng orcid.org/0000-0002-8716-55581 na1, Alexander Kukalev1 na1, Izabela Harabula1,2 na1, Luna Zea-Redondo1,2 na1, Dominik Szabó1,2 na1, Mandy Meijer orcid.org/0000-0003-3314-12243, Leonid Serebreni1 nAff16, Yingnan Zhang4, Simona Bianco orcid.org/0000-0001-5819-060X5, Andrea M. Chiariello orcid.org/0000-0002-6112-01675, Ibai Irastorza-Azcarate1, Christoph J. Thieme orcid.org/0000-0002-1566-09711, Thomas M. Sparks1, Sílvia Carvalho1,6,7,8, Luca Fiorillo orcid.org/0000-0003-2967-01235, Francesco Musella5, Ehsan Irani1,9, Elena Torlai Triglia orcid.org/0000-0002-6059-01161 nAff17, Aleksandra A. Kolodziejczyk10,11 nAff18, Andreas Abentung12 nAff19, Galina Apostolova12, Eleanor J. Paul orcid.org/0000-0003-1183-928513 nAff20 nAff21, Vedran Franke orcid.org/0000-0003-3606-679214, Rieke Kempfer1,2, Altuna Akalin orcid.org/0000-0002-0468-011714, Sarah A. Teichmann orcid.org/0000-0002-6294-636610,11, Georg Dechant12, Mark A. Ungless13, Mario Nicodemi5,9, Lonnie Welch4, Gonçalo Castelo-Branco orcid.org/0000-0003-2247-93933,15 & Ana Pombo orcid.org/0000-0002-7493-62881,2,9 Nature (2021)Cite this article The three-dimensional (3D) structure of chromatin is intrinsically associated with gene regulation and cell function1,2,3. Methods based on chromatin conformation capture have mapped chromatin structures in neuronal systems such as in vitro differentiated neurons, neurons isolated through fluorescence-activated cell sorting from cortical tissues pooled from different animals and from dissociated whole hippocampi4,5,6. However, changes in chromatin organization captured by imaging, such as the relocation of Bdnf away from the nuclear periphery after activation7, are invisible with such approaches8. Here we developed immunoGAM, an extension of genome architecture mapping (GAM)2,9, to map 3D chromatin topology genome-wide in specific brain cell types, without tissue disruption, from single animals. GAM is a ligation-free technology that maps genome topology by sequencing the DNA content from thin (about 220 nm) nuclear cryosections. Chromatin interactions are identified from the increased probability of co-segregation of contacting loci across a collection of nuclear slices. ImmunoGAM expands the scope of GAM to enable the selection of specific cell types using low cell numbers (approximately 1,000 cells) within a complex tissue and avoids tissue dissociation2,10. We report cell-type specialized 3D chromatin structures at multiple genomic scales that relate to patterns of gene expression. We discover extensive ‘melting’ of long genes when they are highly expressed and/or have high chromatin accessibility. The contacts most specific of neuron subtypes contain genes associated with specialized processes, such as addiction and synaptic plasticity, which harbour putative binding sites for neuronal transcription factors within accessible chromatin regions. Moreover, sensory receptor genes are preferentially found in heterochromatic compartments in brain cells, which establish strong contacts across tens of megabases. Our results demonstrate that highly specific chromatin conformations in brain cells are tightly related to gene regulation mechanisms and specialized functions. To explore how genome folding is related to cell specialization, we applied immunoGAM to mouse brain tissue slices and analysed three cell types with diverse functions (Fig. 1a): oligodendroglia (oligodendrocytes and their precursors (OLGs)) from the somatosensory cortex; pyramidal glutamatergic neurons (PGNs) from the cornu ammonis 1 (CA1) of the dorsal hippocampus; and dopaminergic neurons (DNs) from the ventral tegmental area (VTA) of the midbrain. OLGs are important for neuronal myelination and circuit formation11, whereas PGNs are important for temporal and spatial memory formation and consolidation12, and DNs are activated during cue-guided reward-based learning13. Publicly available GAM data from mouse embryonic stem (mES) cells9 were used for comparison (Supplementary Table 1).Fig. 1: ImmunoGAM captures cell-type-specific chromatin contacts in the mouse brain.a, ImmunoGAM was applied to three brain cell types: OLGs, DNs and PGNs (one independent biological replicate for OLGs and two replicates for DNs and PGNs). b, Schematic of the ImmunoGAM workflow. OLGs were selected by immunolabelling with GFP, DNs with tyrosine hydroxylase and PGNs using tissue morphology. Nuclear profiles were laser microdissected, each from a single cell, with three collected together, as described for multiplex-GAM9. c, Example of cell-type-specific contact differences at the Pcdh locus (chromosome 18: 36–39 Mb). GAM matrices represent co-segregation frequencies of 50-kb genomic windows using normalized pointwise mutual information (NPMI). Dashed lines illustrate cell-type differences. NPMI scales range between 0 and 99th percentile per cell type. Contact density heatmaps represent insulation scores using 100–1,000 kb square sizes. RNA-seq and ATAC-seq tracks represent normalized pseudobulk reads from scRNA-seq and scATAC-seq, respectively, except for bulk ATAC-seq from mES cells. d, Strong contacts between Vmn and Olfr receptor gene clusters on chromosome 17 (0–60 Mb) within B compartments (Comp.), separated by ~35 Mb, are observed in brain cells but not in mES cells. Compartments A and B were classified using normalized PCA eigenvectors2.Source dataWe selected cell types from brain tissue slices by immunofluorescence with cell marker antibodies before genomic extraction (Fig. 1b). A detailed flowchart of immunoGAM quality control (QC) measures and normalization is shown in Extended Data Fig. 1a–d and Supplementary Table 2. GAM contact matrices, each from about 850 cells, had low biases in GC content and mappability (Extended Data Fig. 2a–c). We calculated local contact densities and topological domains using the insulation square method14, and calculated compartments associated with open chromatin (compartment A) and closed chromatin (compartment B) using principal component analysis (PCA)2 (Supplementary Tables 3–5).As an example of cell-type-specific organization, we considered the Pcdh locus, which contains three clusters of cell adhesion genes (Pcdha, Pcdhb and Pcdhg) and occupies two topologically associating domains (TADs) in mES cells, as previously described15 (Fig. 1c, see Extended Data Fig. 3a for replicates). Mapping contact densities using 100–1,000 kb insulation squares showed that the locus is generally open above 500 kb. Higher expression of Pcdha and Pcdhb coincides with increased long-range contacts between the three clusters in neurons16 and OLGs17 and with additional long-range contacts with the highly expressed Fgf1 gene in OLGs. We also discovered contacts spanning tens of megabases in brain cells. For example, strong contacts connected two regions approximately 3- and 5-Mb wide, separated by 35 Mb, which contained clusters of vomeronasal (Vmn) and olfactory (Olfr) receptor genes (Fig. 1d, see Extended Data Fig. 3b for replicates). Thus, the application of immunoGAM in specific brain cell types reveals large rearrangements in 3D chromatin architecture at short-range and long-range genomic lengths.To further investigate how cell-type-specific 3D genome topologies relate to gene expression and chromatin accessibility, we produced or collected published single-cell RNA sequencing (scRNA-seq) data and single-cell assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) data from mES cells, the cortex, the hippocampus and the midbrain (Methods, Extended Data Fig. 4, Supplementary Table 6). After selecting cell populations equivalent to those captured by immunoGAM, we compiled cell-type-specific pseudobulk RNA-seq and ATAC-seq datasets.Complex and extensive cell-type-specific changes in TAD-level contacts were frequent, for example, at a 4-Mb region that contains Scn genes that encode sodium voltage-gated channel subunits (Fig. 2a, see Extended Data Fig. 5a for replicates). We obtained a total of approximately 2,300 TADs across cell types, with a median length of about 1 Mb, which is in line with previous reports6 (Extended Data Fig. 5b). Although pairwise comparisons of TAD border positions confirmed previous levels of conservation4,6 (78–89%; Extended Data Fig. 5c), multiway comparisons showed high cell-type specificity (Fig. 2b, see Extended Data Fig. 5d for sparser combinations). One-third of the borders were unique and significantly more insulated in other cell types (Extended Data Fig. 5e), with some variability noted between biological replicates (59–65%) (Extended Data Fig. 5f). By contrast, only 8% of the total set of borders was shared by brain cells and 14% by all cell types. Shared borders showed significantly stronger insulation in brain cells than in mES cells (Extended Data Fig. 5g), which suggests that there is structural stabilization after terminal differentiation. Unique boundaries often contained expressed genes (52–55% in brain cells, 38% in mES cells) (Extended Data Fig. 5h) and genes with enriched Gene Ontology (GO) terms relevant to the specialized cell type (Fig. 2c, Supplementary Table 7), such as ‘membrane depolarization’ and ‘cognition’ in PGNs or genes important for dopaminergic differentiation and dopamine synthesis in DNs.Fig. 2: Chromatin domains rearrange extensively in brain cells, notably at long genes that undergo melting events.a, Example of cell-type-specific contacts at genomic regions (chromosome 2: 64.3–67.3 Mb) with differential expression. Dashed boxes represent 500 kb insulation scores used to determine TAD boundaries (indicated with coloured boxes below). Replicate 1 is shown for brain cells. b, UpSet plots representing multiway TAD boundary comparisons show extensive cell-type specificity. Boundaries were defined as 150 kb genomic regions centred on the lowest insulation score windows and were considered different when separated by >50 kb edge-to-edge. c, Cell-type-specific borders contain genes with GO terms relevant for cell functions. The top four GO terms were the most enriched, and the fifth was selected (over-representation measured by Z-score; one-sided Fisher’s exact permuted P values 300 kb, 479 genes) to determine melting scores from contact density maps that represent insulation score values using 100–1,000 kb squares. Genes were considered to melt if the melting score computed across their coding region was >5 (P < 1 × 10−5; one-sided Kolmogorov–Smirnov testing using maximum distances between distributions). g, Melting associates with higher expression, especially in PGNs and DNs (two-sided Wilcoxon rank-sum test; **P < 0.01, ****P 300 kb) and produce many isoforms owing to complex RNA processing18. Chromatin reorganization was most apparent at long genes in both PGNs and DNs (Fig. 2d, e). For example, Grik2 loses contact density in PGNs compared to mES cells, especially around the transcription start site (TSS) and transcription end site (TES) (Fig. 2d). By contrast, Dscam decondenses across its entire gene body in DNs (Fig. 2e). To assess whether decondensation relates to the expression of long genes, we compared the insulation of the most and least expressed long genes (Extended Data Fig. 5i). Highly expressed genes were significantly less insulated at TSSs and TESs and throughout gene bodies in both DNs and PGNs, but not in OLGs or mES cells. The general contact loss at highly expressed long neuronal genes is reminiscent of the decondensation, or ‘melting’, observed by microscopy at polytene chromosome puffs19 or tandem gene arrays20.To detect melting genome-wide in an unbiased manner, we devised the MELTRON pipeline. MELTRON calculates a ‘melting score’ as the significant difference between cumulative probabilities of insulation scores across a range of genomic scales (100–1,000 kb) between two cell types and within regions of interest, here defined as all (479) long genes (Fig. 2f). We found 120–180 melting genes with melting scores of >5 (Kolmogorov–Smirnov test, P < 1 × 10−5) between brain cells and mES cells (Fig. 2g, Supplementary Table 8). Grik2 had melting scores of 12 and 26 in PGNs (replicates 1 and 2, respectively), whereas Dscam had scores of 38 and 50 in DNs (replicates 1 and 2, respectively) and Magi2 had a score of 73 in OLGs (Extended Data Fig. 6a, b). Melting scores in the PGN and DN replicates correlated well (Extended Data Fig. 6c).Melting genes were significantly more transcribed and showed higher chromatin accessibility than non-melting long genes, especially in PGNs and DNs (Fig. 2g, Extended Data Fig. 6d–f). Of interest, many top (3%) melting genes (24 out of 44) are sensitive to topoisomerase I inhibition in ex vivo neuronal cultures21, which was in contrast to 16% (42 out of 261) with intermediate melting scores or 16% of non-melting genes (Extended Data Fig. 6g). This result suggests that extensive melting of long genes is associated with the resolution of topological constraints21. Meltinggenes often belonged to compartment A in both mES cells and the corresponding brain cell (43–58%), especially when highly transcribed in both cell types (Extended Data Fig. 6h)
https://www.nature.com/articles/s41586-021-04081-2
Cell-type specialization is encoded by specific chromatin topologies