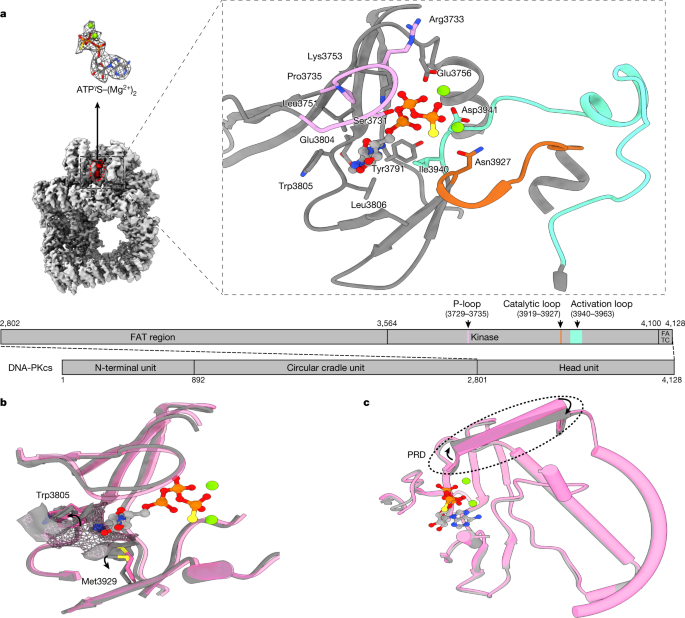
Structural insights into inhibitor regulation of the DNA repair protein DNA-PKcs The DNA-dependent protein kinase catalytic subunit (DNA-PKcs) has a central role in non-homologous end joining, one of the two main pathways that detect and repair DNA double-strand breaks (DSBs) in humans1,2. DNA-PKcs is of great importance in repairing pathological DSBs, making DNA-PKcs inhibitors attractive therapeutic agents for cancer in combination with DSB-inducing radiotherapy and chemotherapy3. Many of the selective inhibitors of DNA-PKcs that have been developed exhibit potential as treatment for various cancers4. Here we report cryo-electron microscopy (cryo-EM) structures of human DNA-PKcs natively purified from HeLa cell nuclear extracts, in complex with adenosine-5′-(γ-thio)-triphosphate (ATPγS) and four inhibitors (wortmannin, NU7441, AZD7648 and M3814), including drug candidates undergoing clinical trials. The structures reveal molecular details of ATP binding at the active site before catalysis and provide insights into the modes of action and specificities of the competitive inhibitors. Of note, binding of the ligands causes movement of the PIKK regulatory domain (PRD), revealing a connection between the p-loop and PRD conformations. Electrophoretic mobility shift assay and cryo-EM studies on the DNA-dependent protein kinase holoenzyme further show that ligand binding does not have a negative allosteric or inhibitory effect on assembly of the holoenzyme complex and that inhibitors function through direct competition with ATP. Overall, the structures described in this study should greatly assist future efforts in rational drug design targeting DNA-PKcs, demonstrating the potential of cryo-EM in structure-guided drug development for large and challenging targets. DNA double-strand breaks (DSBs) are the most toxic form of DNA damage. Two major pathways, homologous recombination (HR) and non-homologous end joining (NHEJ), repair DSBs1. HR requires DNA end resection and is active during the S and G2 phases of the cell cycle, when a sister chromatid is available as a repair template5. By contrast, NHEJ directly ligates DNA ends efficiently in the absence of any template2.DSBs lead to increased genome instability and trigger cell death. This is widely exploited in the treatment of cancer in both radiotherapy using ionizing radiation (IR) and chemotherapy using topoisomerase II inhibitors2,3. However, intrinsic DNA damage response and repair provide tumour cells with some protection. The pathological DSBs caused by IR and topoisomerase II inhibitors are mainly repaired by NHEJ, which requires DNA-dependent protein kinase (DNA-PK), comprising the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and a Ku70–Ku80 heterodimer6,7,8. DNA-PKcs, which belongs to the phosphoinositide 3-kinase (PI3K)-related protein kinase (PIKK) family, has a central role in the regulation of NHEJ9. In combination with IR or genotoxic chemotherapy, inhibition of DNA-PK kinase activity can improve cancer therapy4. There have been many efforts to develop small-molecule inhibitors targeting the ATP-binding site of DNA-PKcs, informed by early studies of synthetic small-molecule PI3K inhibitors10. The older-generation DNA-PK inhibitors, including CC115, KU-0060648, LY294002, LY3023414, NU7026 and NU7441, are effective, but all have different limited selectivity against PI3K and PIKK members (especially mTOR and PI3Kγ). The newer-generation inhibitors, developed from large-scale screening, include VX-984, M3814 and AZD7648, which have better selectivity for DNA-PKcs4,10,11.So far, to our knowledge, no structures have been published of DNA-PKcs in complex with ATP or any inhibitor. This has limited understanding of both ATP binding to DNA-PKcs and the mode of action of the inhibitors, including drug candidates, and has posed a major hurdle to lead development. Here we report structures of DNA-PKcs in complex with adenosine-5′-(γ-thio)-triphosphate (ATPγS) and other inhibitors, including the broad-spectrum PI3K inhibitor wortmannin, the older-generation DNA-PKcs selective inhibitor NU7441 and the newer-generation AZD7648 and M3814, which are in clinical trials. The structures allow understanding of DNA-PKcs binding to ATP before substrate binding and catalysis, the modes of action of the inhibitors and the mechanisms by which they achieve specificity. They also provide structural guidance for future drug design targeting DNA-PKcs, demonstrating the great potential of cryo-EM in structure-based drug discovery.DNA-PKcs purified from HeLa cell nuclear extract was incubated with ATPγS and loaded onto a previously prepared grid with a support film of homemade single-layer graphene oxide (Extended Data Fig. 1). The overall resolution of the DNA-PKcs–ATPγS complex is 3.40 Å with the local resolution of the catalytic core region better than 3 Å, showing clear density for the modelling of ATPγS with two Mg2+ ions and demonstrating a similar structure to that of the homologue mTOR (Fig. 1a and Extended Data Fig. 2a)12. The p-loop (residues 3729–3735) interacts closely with the phosphate groups of ATPγS. Central to this is the interaction between the β-phosphate and the main chain NH group of Arg3733, together with the side chain of Ser3731. Lys3753 interacts with the α-phosphate of ATPγS. Asn3927, Asp3941 and Glu3756 together coordinate the Mg2+ ions. The γ-phosphate of ATPγS points towards the substrate-binding site. The ‘hinge loop’ between the N-lobe and C-lobe, together with the two lobes, constitutes a hydrophobic surface, formed by the side chains of Leu3751, Tyr3791, Trp3805, Leu3806 and Ile3940 and the main chain of Glu3804, to which the adenine moiety of ATPγS can bind. Thus, ATPγS and the Mg2+ ions bind in the ATP-binding groove of DNA-PKcs, coordinating the N- and C-lobes, including the p-loop, activation loop and catalytic loop of the kinase. Conformational changes include the outward rotation of both Trp3805 and Met3929, opening the pocket to accommodate ATP (Fig. 1b)13, and coordinated movements of α-helices in the head unit of the protein (Fig. 1c). Notably, the PRD (residues 4009–4039) does not interfere with the interaction of DNA-PKcs with ATP and Mg2+. However, the α-helix of the PRD (residues 4009–4023) of DNA-PKcs is moved away from the substrate-binding site following ATP binding, partly relieving the blockage by PRD of the substrate-binding site required for subsequent catalysis (Fig. 1c).Fig. 1: ATPγS–(Mg2+)2 interaction with and regulation of DNA-PKcs.a, Coulomb potential map of the DNA-PKcs–ATPγS–(Mg2+)2 complex. The expanded view shows ATPγS–(Mg2+)2 binding in the ATP-binding groove. ATPγS (light grey), together with two Mg2+ ions (fluorescent green), coordinates the N- and C-lobes, especially the p-loop (plum), catalytic loop (chocolate) and activation loop (azure) of DNA-PKcs (grey). The γ-phosphate group points towards the substrate-binding site. The top left image shows the clear Coulomb potential map for modelling of ATPγS–(Mg2+)2, while the schematic representation below highlights the three units of DNA-PKcs and detailed composition of the head unit. b, Opening of the ATP-binding groove entrance. The residues on both sides of the ATP-binding groove entrance, Trp3805 and Met3929, exhibit an outward rotation that allows docking of the adenine moiety of ATPγS. Apo DNA-PKcs (Protein Data Bank (PDB), 6ZFP) is coloured pink with a mesh surface. c, ATPγS–(Mg2+)2 regulation on the PRD. PRD blocks the substrate-binding site. When ATP binds, PRD is tilted and moves away from its position in the apo structure.DNA-PKcs was incubated with several DNA-PKcs inhibitors with different specificities (wortmannin, NU7441, AZD7648 and M3814), to investigate protein–ligand interactions (Supplementary Table 1). The overall resolutions of the structures range from 2.96 Å to 3.33 Å. In all the structures, the kinase core has the highest local resolution, allowing the inhibitors to be unequivocally modelled (Fig. 2a).Fig. 2: ATP competitive inhibitors (wortmannin, NU7441, AZD7648 and M3814) and their modes of binding to DNA-PKcs.a, Inhibitors investigated and their corresponding Coulomb potential maps. b, Binding of wortmannin (green) to the ATP-binding site, where it is covalently modified by the primary amine group of Lys3753. c, Binding of NU7441 (blue) to the ATP-binding site. d, Binding of AZD7648 (purple) to the ATP-binding site. e, Binding of M3814 (cyan) to the ATP-binding site. DNA-PKcs is shown in grey.Wortmannin is one of the early DNA-PK inhibitors used for kinase inhibition14. In the cryo-EM structure, wortmannin packs on one side, mainly against the N-lobe of DNA-PKcs (Leu3751, Ile3803 and Trp3805), while on the other side it packs against the C-lobe (Met3929 and Asp3940) (Fig. 2b). One edge of the compound is facing the deep pocket of the ATP-binding site, and the opposite edge is exposed to solvent. The primary amine (ε-amino group) of Lys3753 forms a covalent C–N bond with C19 in the furan ring of wortmannin (Fig. 2a), irreversibly inhibiting kinase activity. The ATP-binding site of DNA-PKcs has close structural complementarity to wortmannin. The C14 methyl resides in a pocket formed by Leu3751, Ile3803 and Trp3805, while the C10 methyl fits into a pocket formed by Met3729, Pro3735 and Leu3751 (Fig. 2b). There is no density corresponding to the acetoxy group attached to C11 of wortmannin, as was observed in the published structure of wortmannin in complex with porcine PI3Kγ (Extended Data Fig. 3a)15.Compared with wortmannin, NU7441 is a more selective inhibitor, developed from the earlier inhibitors NU7026 and LY294002, which were derived from the broad-spectrum protein kinase flavonoid inhibitor quercetin16. The chromone core and morpholino ring of NU7441 bind in the centre of the deepest pocket formed by Leu3751, Tyr3791, Ile3803, Trp3805, Leu3806, Ile3940 and Met3929, within which O1 and O27 of NU7441 form hydrogen bonds with the peptide backbones of Asp3941 and Leu3806 (Fig. 2c). The dibenzothiophene group interacts with the N-lobe and docks in the hydrophobic groove of Met3729, Pro3735 and Leu3751 (Fig. 2c).AZD7648 is a recently developed DNA-PK inhibitor shown to be highly selective and to enhance the efficacy of doxorubicin and IR11,17. Moreover, AZD7648 was identified as a potential combinational therapy with the PARP inhibitor olaparib and is currently under clinical trial for advanced malignancies (trial identifier, NCT03907969)11. The developers of AZD7648 took advantage of a previously published crystal structure of AZD7648 bound to PI3Kγ17. In the DNA-PKcs–AZD7648 complex, the compound binds the hinge loop (Fig. 2d). The triazolopyridine moiety with a methyl group lies in the deep hydrophobic pocket formed by Tyr3791, Ile3803, Leu3806 and Ile3940. The purine moiety docks in the narrow tunnel formed by Trp3805, Leu3806 and Met3929. Similarly to the PI3Kγ–AZD7648 complex, the N3 hydrogen of AZD7648 (the aniline NH) forms bonds with the backbone oxygen of Glu3804 and N7 accepts the hydrogen from the backbone NH group of Leu3806 (ref. 17). N6 in the triazolopyridine moiety also binds to the backbone NH group of Asp3941. There is a further hydrogen bond in the case of DNA-PKcs–AZD7648: N5 forms a bond with the primary amine group of Lys3753. Comparison of the two structures shows a 90° rotation of the indole ring for Trp812 in PI3Kγ relative to Trp3805 in DNA-PKcs, which in DNA-PKcs not only provides a better hydrophobic binding surface but also aligns nicely to the AZD7648 purine ring, leading to a π stacking interaction that enhances affinity (Extended Data Fig. 3b).M3814 (peposertib or nedisertib) is another recently developed DNA-PK selective inhibitor18,19. Preclinical studies have revealed its synergy with radiotherapy and chemotherapies using doxorubicin and etoposide20. The compound was shown to be well tolerated with modest efficacy in unselected tumours in phase 1 clinical trial results (trial identifier, NCT02316197)19; M3814 is currently in four phase 2 clinical trials targeting different cancers (trial identifiers, NCT03770689, NCT04068194, NCT04071236 and NCT04172532). According to our structure, the quinazoline and morpholino moieties of the compound fit well into the deepest, largely hydrophobic pocket formed by the side chains of Leu3751, Tyr3791, Val3793, Ile3803, Trp3805, Leu3806, Ile3938 and Ile3940 and the main chains of Glu3804 and Asp3941 (Fig. 2e). The chloro-fluorobenzene ring rotates by ~60° and points towards the N-lobe, facing the side chains of Met3729, Ser3731, Pro3735, Leu3751 and Lys3753. The remaining moiety containing the pyridazine ring then rotates back to be almost parallel to the quinazoline plane, lying in the groove of Met3729, Trp3805, Thr3811, Asn3926 and Met3929.All the inhibitors studied target the ATP-binding groove of DNA-PKcs, overlapping with the ATPγS-binding site (Fig. 3). Among them, wortmannin has the maximum overlap. Compared with ATPγS, wortmannin has greater complementarity to the binding site (Fig. 3a), binding deeper into the ATP adenine-moiety pocket, with its two protruding methyl groups fitting better into the hydrophobic pocket on the surface of the N-lobe.Fig. 3: Comparisons of the binding modes among ATPγS–(Mg2+)2 and the four inhibitors.a, Comparison of the binding modes of ATPγS–(Mg2+)2 and wortmannin in DNA-PKcs. Top, binding conformations of ATPγS–(Mg2+)2 (light grey, ATPγS; fluorescent green, Mg2+ ions) and wortmannin (green). Bottom, conformational differences in the binding groove of DNA-PKcs between ATPγS–(Mg2+)2 (grey) and wortmannin (green). b, Comparison of the binding modes of ATPγS–(Mg2+)2 and NU7441 in DNA-PKcs. Top, binding conformations of ATPγS–(Mg2+)2 and NU7441 (blue). Bottom
https://www.nature.com/articles/s41586-021-04274-9
Structural insights into inhibitor regulation of the DNA repair protein DNA-PKcs