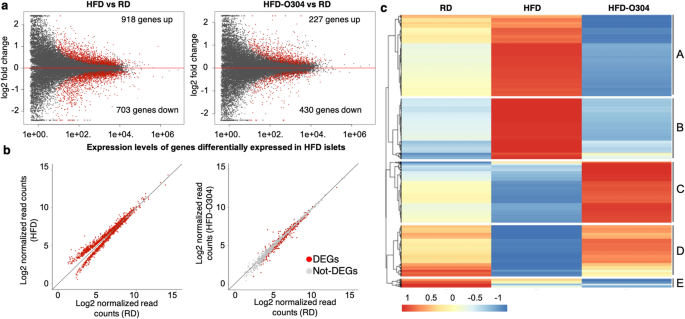
Pan-AMPK activator O304 prevents gene expression changes and remobilisation of histone marks in islets of diet-induced obese mice AMP-activated protein kinase (AMPK) has an important role in cellular energy homeostasis and has emerged as a promising target for treatment of Type 2 Diabetes (T2D) due to its beneficial effects on insulin sensitivity and glucose homeostasis. O304 is a pan-AMPK activator that has been shown to improve glucose homeostasis in both mouse models of diabetes and in human T2D subjects. Here, we describe the genome-wide transcriptional profile and chromatin landscape of pancreatic islets following O304 treatment of mice fed high-fat diet (HFD). O304 largely prevented genome-wide gene expression changes associated with HFD feeding in CBA mice and these changes were associated with remodelling of active and repressive chromatin marks. In particular, the increased expression of the β-cell stress marker Aldh1a3 in islets from HFD-mice is completely abrogated following O304 treatment, which is accompanied by loss of active chromatin marks in the promoter as well as distant non-coding regions upstream of the Aldh1a3 gene. Moreover, O304 treatment restored dysfunctional glucose homeostasis as well as expression of key markers associated with β-cell function in mice with already established obesity. Our findings provide preclinical evidence that O304 is a promising therapeutic compound not only for T2D remission but also for restoration of β-cell function following remission of T2D diabetes. The current global epidemic of Type 2 Diabetes (T2D) poses a major health threat and is strongly associated with the worldwide rise in obesity1. Exercise and controlled diets improve insulin sensitivity and are the first treatment recommendations for T2D patients, but long-term compliance is difficult and for some patients exercise and diet does not suffice or are not feasible options. Notably, accumulating data show that, provided that β-cell function has not deteriorated too far2, remission of T2D with restoration of β-cell function can be achieved following a very low calorie diet3, intensive insulin therapy4 and bariatric surgery5. However, how remission of T2D diabetes, e.g. following caloric restriction, affects the β-cell on a molecular level is largely unknown. A key mediator of the effects of exercise and diet/caloric restriction on glucose metabolism is AMP-activated protein kinase (AMPK)6. AMPK, the key sensor of cellular energy homeostasis, is activated under conditions of low energy and phosphorylates targets in numerous signalling pathways, including lipid homeostasis, mitochondrial biogenesis, and glycolysis, thus favouring catabolic processes at the expense of anabolism7. Given its important role in cellular energy homeostasis and positive effects on insulin sensitivity, AMPK has emerged as a promising target for treatment of T2D.We recently described a novel pan-AMPK activator, O304, that exerts beneficial effects on glucose homeostasis and vascular health both in high-fat diet (HFD) induced obese mice and in human T2D subjects8. Importantly, O304 treatment preserved β-cell function and secretory capacity by alleviating metabolic stress, largely through increased glucose uptake in the peripheral tissues thus improving insulin sensitivity and consequently promoting β-cell rest8. Notably, direct effects of O304 in alleviating ER stress and inhibiting amyloid formation in β-cells were also demonstrated in ex vivo cultured islets8. The β-cell molecular consequences following O304 mediated in vivo restoration of glucose homeostasis are however not fully understood. In particular, genome-wide changes in the transcriptional profile of pancreatic islets, as well as potential changes in genome-wide epigenetic chromatin marks, have not been assessed in the context of O304-mediated remission of insulin resistance and dysfunctional glucose homeostasis. To study the effects of O304 mediated amelioration of insulin resistance and dysglycemia on the transcriptional landscape and epigenetic modifications in pancreatic islets within an in vivo prediabetic context, we here describe the analyses of transcriptomes as well as chromatin marks in pancreatic islets of O304 treated HFD-mice. We show that O304 treatment largely prevented islet gene expression changes associated with HFD-induced obesity in pancreatic islets and preserved the distribution of epigenetic signatures associated with active or repressed chromatin. Furthermore, O304 treatment restored glucose homeostasis and gene expression levels of key markers involved in β-cell stress in islets of HFD-induced obese mice.We have previously demonstrated that the AMPK activator O304 improves blood glucose homeostasis in both human T2D subjects as well as in high-fat diet induced obese and diabetic mouse models. In the present study, we have now analysed the in vivo effects of O304 treatment on the genome-wide transcriptional program of the pancreatic islets. To characterize gene expression signatures and chromatin marks of β-cells in the context of elevated glucose levels induced by obesity, and the potential positive effects of O304-mediated remission of dysglycemia in this context, we performed RNA-sequencing and Chromatin ImmunoPrecipitation sequencing (ChIP-seq) of islets isolated from 20-week-old mice on regular diet (RD), high-fat diet (HFD) and HFD formulated with O304 (HFD-O304). Diets were introduced at 10 weeks of age. Fasting blood glucose and plasma insulin levels were similar between the groups at start of the diet (Supplementary Fig. S1a). In line with our previous data8, HFD-O304 mice had significantly lower glucose and insulin levels than HFD mice after 9 weeks of the treatment period (Supplementary Fig. S1b). Food intake was highest for RD mice (4.2 g/m/day) and lowest for HFD mice (2.5 g/m/day), whereas food intake was increased in HFD-O304 mice compared to HFD fed mice (3.2 g/m/day, Supplementary Fig. S1c). A blunted first-phase insulin release is the first detectable defect in β-cell function in T2D aetiology and can be assessed by arginine stimulated insulin secretion tests, which provides an estimate of the β-cell secretory capacity independent of potential increased peripheral glucose uptake/improved insulin. Arginine stimulated insulin secretion was improved in HFD-O304 mice as compared to non-treated HFD mice (Supplementary Fig. S1d), confirming previous data on improved first-phase insulin secretory capacity of the β-cells in O304 treated diet-induced obese mice8.RNA-seq and ChIP-seq analysis of islets isolated from mice following 10 weeks of treatment demonstrated that 1621 genes were differentially regulated in islets of HFD mice compared with that of RD mice (918 upregulated, 703 downregulated: FDR 90% of the differentially expressed genes in islets of HFD mice (Fig. 1b). Accordingly, both principal component analysis (PCA) (Supplementary Fig. S2b) and hierarchical clustering (Supplementary Fig. S2c) group HFD-O304 islet samples closer to islets from RD controls than to islets of HFD mice. Taken together, these findings provide evidence that O304 treatment prevents HFD induced islet transcriptional changes, thus largely preserving transcriptional profiles similar to islets of RD mice.Figure 1O304 treatment prevents islet gene expression changes induced by HFD in mice. (a) MA plot of gene expression in HFD islets versus RD islets (left) and HFD-O304 islets versus RD-islets (right). Red dots indicate differentially expressed genes (FDR < 0.01). (b) Gene expression levels of differentially expressed genes between HFD islets and RD islets (left) and the same genes plotted between HFD-O304 islets and RD islets (right). Red dots indicate genes that are differentially expressed in each condition (FDR < 0.01). Note that the vast majority of genes differentially expressed in HFD islets are expressed at normal levels in HFD-O304 islets. (c) K-means clustering of genes differentially expressed between RD (left), HFD (middle), and HFD-O304 islets (right). Scale from red to blue indicates fold change over average read count for each row.K-means clustering analysis of differentially expressed genes revealed 4 major clusters, out of a total of 5, where clusters A and B contain genes primarily upregulated in islets of HFD mice, and clusters C and D contain genes primarily downregulated in islets from HFD mice (Fig. 1c). Enrichment analysis of gene ontology (GO) terms and KEGG pathways revealed profound functional differences between the expression profiles of the three experimental groups (Supplementary Fig. S3 and Supplementary Table S1). The most enriched GO- and KEGG-terms for cluster A (containing genes that were upregulated in HFD islets) are related to endoplasmatic reticulum (ER) function and protein processing, including response to ER stress (Supplementary Fig. S3, Supplementary Table S1). This finding corroborates our previous data on ex vivo cultured islets where O304 treatment prevents increased expression of genes related to unfolded protein response under high-glucose conditions8. In the setting of obesity-provoked insulin resistance, β-cell proliferation is a compensatory response to the increased demand of insulin. Consistently, upregulation of genes in cluster B, which are mainly related to cell cycle and proliferation (Supplementary Fig. S3, Supplementary Table S1), were observed in islets of HFD mice, but not in islets of HFD-O304 mice. β-Cell proliferation has also been suggested to be tightly coupled to a loss of mature β-cell identity21, and AMPK has been implicated in maintenance of a mature β-cell phenotype via inhibition of mTORC1 signalling22. Still, while β-cell proliferative markers were not induced in islets of HFD-O304 mice (Supplementary Fig. S4a), we find no clear evidence for altered mTORC1 signalling (Supplementary Fig. S4b). Taken together these findings show that O304 treatment prevents up-regulation of genes associated with metabolic β-cell stress. However, given that O304 averts HFD induced insulin resistance8 (Supplementary Fig. S1), the effect of O304 on β-cell stress is likely partly indirect.Genes in Cluster C, which were most abundantly expressed in islets of HFD-O304 mice and least expressed in HFD mice, were strongly enriched for terms/pathways related to regulation of hormonal secretion and insulin secretion in particular (Supplementary Fig. S3, Supplementary Table S1), as well as genes related to AMPK- and glucagon-signalling (Supplementary Table S1). Compared to islets from RD mice, O304 treatment preserved or even increased the expression of these genes in mice fed HFD (Supplementary Fig. S3, Supplementary Table S1). These findings suggest that, in the context of HFD, O304 treatment enhances the insulin secretory capacity of β-cells by increasing the abundance of transcripts related to these processes. Cluster D was enriched in genes largely related to neuronal function, which may reflect that β-cells share several phenotypic traits with neurons that are dependent on similar transcriptional programs for their function23. Last, cluster E genes are mainly associated with amino acid metabolism and other catabolic processes (Supplementary Fig. S3), likely reflecting that AMPK activation inhibits protein synthesis and stimulates catabolic processes24.Taken together, these data demonstrate that O304 treatment largely prevented the transcriptional changes observed in islets of HFD mice. Although the net action of O304 treatment on β-cells likely occurs indirectly via amelioration of insulin resistance and dysglycemia as well as through a combination of direct downstream targets, it significantly impacts on several key β-cell processes affected in T2D aetiology, and in particular O304 treatment prevented gene expression changes associated with insulin signalling and hormonal secretion.O304 treatment mitigates gene expression signatures associated with β-cell stressTo further understand the impact of O304 treatment on glucose homeostasis and β-cell function in the context of HFD and insulin resistance, we explored our transcriptional dataset to investigate the effects on specific genes relevant for β-cell function and/or β-cell identity, and whose altered expression has been implicated in β-cell stress. As HFD mice are not overt diabetic but exhibit a pre-diabetic glucose intolerant phenotype, islets of HFD-mice were expected to display subtle expression changes, primarily of early markers of β-cell stress. Preserved expression levels of such markers would imply a maintained β-cell phenotype, and the functional annotation of differentially expressed genes (Supplementary Fig. S3, Supplementary Table S2) in our data indeed suggest that this may be the case.Aldehyde dehydrogenase 1 isoform A3 (ALDH1A3) is a proposed marker for metabolically stressed β-cells25,26,27,28,29 and in agreement with these findings, Aldh1a3 expression was significantly increased in islets from HFD-mice compared to RD controls (Fig. 2a). Notably, Aldh1a3 expression was reduced in islets of HFD-O304 mice compared with both HFD and RD islets (Fig. 2a). Increased Aldh1a3 expression in β-cells has been linked to reduced expression of key transcription factors associated with maintenance of β-cell identity and function27. No major differences in gene expression were observed for a set of mature β-cell identity genes although the expression profile of HFD-O304 islets were more similar to RD islets than to HFD islets22,30 (Supplementary Fig. S5a). However, O304 treatment not only prevented the reduced expression of NeuroD1 and Isl1 (Fig. 2b,c) observed in HFD islets but also enhanced the expression of these genes compared with that observed in RD islets (Fig. 2b,c). In addition, a similar trend was observed for MafA, the expression of which was slightly decreased in HFD islets (Fig. 2d) as compared to RD and HFD-O304 islets. FoxO1 that regulates NeuroD1 and MafA was slightly upregulated in HFD-O304 islets while O304 treatment had no effect on Pdx1 or Nkx6.1 expression (Fig. 2e–g). The limited transcriptional changes observed for only a subset of β-cell identity genes suggest that increased Aldh1a3 expression is not directly linked
https://www.nature.com/articles/s41598-021-03567-3
Pan-AMPK activator O304 prevents gene expression changes and remobilisation of histone marks in islets of diet-induced obese mice